

Translate this page into:
Classical imaging in cerebral folate transport deficiency

*Corresponding author: Sarbesh Tiwari, Department of Diagnostic and Interventional Radiology, All India Institute of Medical Sciences, Jodhpur, Rajasthan, India. sarbesh1984@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Mahal S, Pamnani J, Parakh M, Tiwari S. Classical imaging in cerebral folate transport deficiency. Case Rep Clin Radiol, doi: 10.25259/CRCR_138_2023
Abstract
Cerebral folate transport deficiency is an inherited disorder with late infantile-onset caused by nonsense mutations in the folate receptor 1 gene coding for folate receptor alpha. We report a case of a 4-year-old female child, who presented with global neurodevelopmental regression with onset at 15 months of age with an electroencephalogram showing poorly modulated polymorphic delta sleep background. Based on the clinical features, age of onset, and imaging findings, the possibility of cerebral folate transport deficiency (OMIM#613068) was considered. Targeted gene sequencing confirmed the diagnosis.
Keywords
Cerebral folate transport deficiency
Hypomyelination
Neurodevelopmental regression

INTRODUCTION
Cerebral folate transport deficiency is an inherited disorder of brain-specific folate transporter caused by nonsense mutations in the folate receptor 1 gene coding for folate receptor alpha. This leads to the introduction of a premature stop codon in messenger ribonucleic acid causing its rapid degradation.[1,2] The syndrome is characterized by late infantile onset, severe developmental regression, epilepsy, hypomyelinating leukodystrophy, and low 5-methyltetrahydrofolate (5-MTHF) concentration in the cerebrospinal fluid (CSF) evaluation. Early diagnosis is critical because folinic acid therapy can potentially reverse the clinical symptoms.
CASE REPORT
A 4-year-old female child with non-consanguineous parents presented with global neurodevelopmental regression starting at 15 months of age. She had associated axial myoclonic seizures 6–7 episodes every day, frequent falls, ataxic gait, and language regression. A detailed neurological examination confirmed the global developmental delay, poor eye contact, incomprehensible sounds, unsteady gait, and hypotonia. The fundus evaluation was normal. The thyroid function test, serum folic acid, and Vitamin B12 levels were within normal limits. An electroencephalogram was done, which showed a poorly modulated polymorphic delta sleep background and the absence of any age-appropriate sleep features. There were pseudo-periodic generalized epileptiform discharges. Magnetic resonance imaging (MRI) brain showed diffuse cerebellar atrophy [Figure 1a]. An abnormal T2 hyperintense signal was noted involving the cerebral subcortical white matter on the T2-weighted image [Figure 1b]. The corresponding areas were hypointense on T1 images [Figure 1c]. These features were suggestive of hypomyelination. Axial susceptibility-weighted imaging showed blooming in the subcortical white matter of the bilateral frontal region and basal ganglia, suggesting calcification [Figure 1d]. Computed tomography of the head [Figure 2] confirmed the presence of symmetric calcification in frontal subcortical white matter and basal ganglia.

- Magnetic resonance imaging brain: Axial T2 weighted image (a) shows diffuse cerebellar atrophy. (b) Axial T2-weighted image at the level of the basal ganglia shows abnormal T2-hyperintense signal involving the cerebral subcortical white matter (yellow arrow), which is inappropriate for age. These areas are hypointense on T1 images (c). These features were suggestive of hypomyelination. (d) Axial susceptibility-weighted imaging shows blooming in the subcortical white matter of the bilateral frontal region (yellow arrows), suggesting calcification.
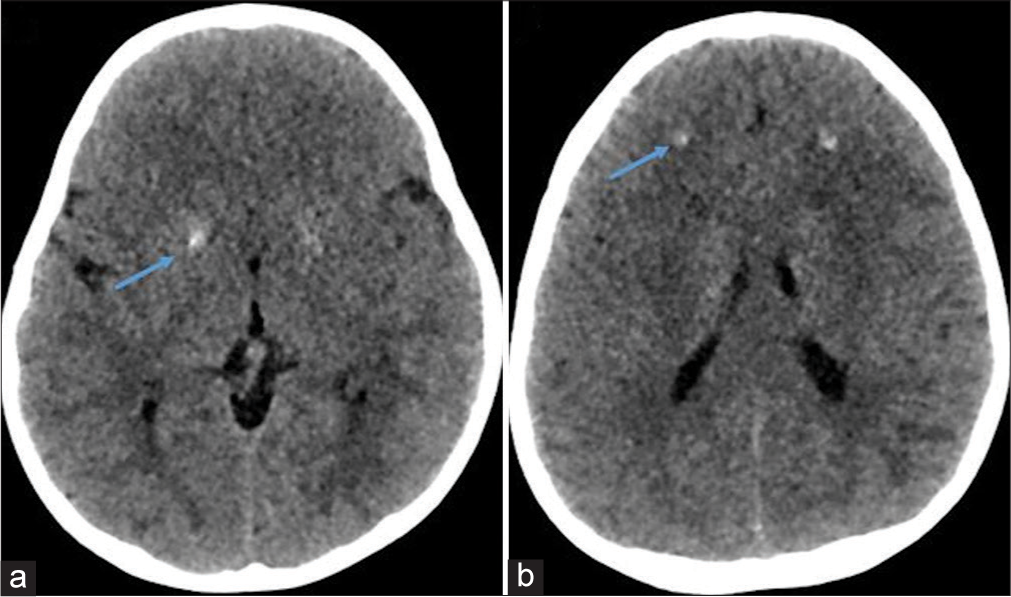
- Computed tomography of the head (blue arrows in a and b) confirmed the presence of symmetric calcification in basal ganglia and frontal subcortical white matter.
Based on the clinical features, age of onset, and imaging findings, the possibility of cerebral folate transport deficiency was considered. Targeted gene sequencing was done, which showed a rare homozygous deletion of single base pair (c. 187 del) in exon 3 of the FOLR1 gene (chr11:g.72195289del; Depth: 139x) that results in frame-shift and truncation of the protein 68 amino acids downstream to the codon 63 (p.Asn63MetfsTer68; ENST00000393679.5), confirming the diagnosis.
DISCUSSION
Mutations in the FOLR1 gene result in a brain-specific folate transport defect characterized by a late infantile-onset of severe developmental regression, sometimes commencing in the 2nd or 3rd year of life. Folate transport into cells is regulated by FOLR1, which is a glycophosphatidylinositol-anchored cell membrane protein. Genetic variants have been found to be responsible for different pathologic variants of this condition, attributing to specific neurological disorders.[2-4] Homozygous mutations and compound heterozygous mutations lead to autosomal recessive disorders, while neurodegeneration is caused by homozygous 18-bp in-frame duplication. In this case, we have added a novel homozygous deletion of a single base pair (c. 187 del) variant in the FOLR1 genotype spectrum.
Almost all cases of cerebral folate transporter deficiency demonstrate significantly low levels of 5-MTHF level in CSF. These children present with refractory myoclonic epilepsy and ataxia, as seen in our patient.[1]
Hypomyelination is a well-established association with this condition. Other common central nervous system abnormalities reported in the literature include cerebral atrophy, focal T2 hyperintense white matter lesions, and diffuse hyperintensities in periventricular, and subcortical white matter on T2-weighted images. The focal T2-hyperintense lesion in periventricular and subcortical white matter is more commonly found in older patients and most probably represents areas of gliosis.[2] Cerebellar atrophy with enlargement of cisterna magna, mild hypoplasia of the vermis, and brainstem atrophy are seen in some cases. Calcification at basal ganglia and the subcortical white matter has been reported in the literature and was also seen in our case.[1] Imaging differentials of subcortical white matter signal changes with calcification include Kearns–Sayre syndrome, Cockayne syndrome, and Aicardi–Goutieres syndrome. The absence of progressive external ophthalmoplegia and cardiac conduction defects were against the diagnosis of Kearns–Sayre syndrome. In Aicardi–Goutières syndrome, calcifications are more frequently punctuate and distributed in the basal ganglia, the deep and periventricular white matter.[5] Cockayne syndrome is characterized by cutaneous photosensitivity, significant white matter loss, and ventricular dilatation, which were not present in our case.[6,7]
Cerebral folate transport deficiency is not exclusively caused by FOLR1 genetic variants. Hereditary folate malabsorption, MTHF, and dihydrofolate reductase deficiency are other significant causes. Normal red blood cell count and plasma concentration of homocysteine and folate within normal limits exclude all these conditions.[2]
It has been reported in several studies that combined oral and intravenous oral folinic acid can improve both clinical symptoms and white matter abnormalities.
CONCLUSION
Cerebral folate deficiency must be kept as differential in central hypomyelination and calcification. This being a potentially treatable inborn error of metabolism, a prompt diagnosis and early initiation of treatment is important to ensure a better outcome.
TEACHING POINTS
Cerebral folate transport deficiency is an inherited neurodegenerative autosomal recessive disorder resulting from brain-specific folate transporter deficiency early in life.
Diagnosis is based on the clinical phenotype of severe developmental regression, epilepsy, progressive ataxia, and reduced 5-methyltetrahydrofolate in CSF analysis.
Magnetic Resonance Imaging (MRI) findings of central hypomyelination with basal ganglia and subcortical calcification in appropriate clinical scenarios should raise a suspicion of cerebral folate transport deficiency.
MCQs
-
Most common age of onset of cerebral folate transporter deficiency is–
Neonatal
Adult onset
Late infantile
Adolescence
Answer Key: c
-
A substrate that is affected in CSF in cases of cerebral folate transporter deficiency is–
Neuregulin 1
P- glycoprotein
Contactin 2
5-methyltetrahydrofolate (5-MTHF)
Answer Key: d
-
The most common clinical feature of cerebral folate transporter deficiency is –
Neuroregression
Refractory epilepsy
Headache
Reduced visual acuity
Answer Key: b
-
Which of the following is not a differential diagnosis of cerebral folate transporter deficiency?
Kearns–Sayre syndrome
Cockayne syndrome
Sturge–Weber syndrome
Aicardi–Goutieres syndrome
Answer Key: c
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The author(s) confirms that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Cerebral folate transporter deficiency syndrome in three siblings: Why genetic testing for developmental and epileptic encephalopathies should be performed early and include the FOLR1 gene. Am J Med Genet A. 2021;185:2526-31.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular characterization of folate receptor 1 mutations delineates cerebral folate transport deficiency. Brain. 2012;135:2022-31.
- [CrossRef] [PubMed] [Google Scholar]
- Progressive ataxia and myoclonic epilepsy in a patient with a homozygous mutation in the FOLR1 gene. J Inherit Metab Dis. 2010;33:795-802.
- [CrossRef] [PubMed] [Google Scholar]
- Cerebral folate deficiency: Analytical tests and differential diagnosis. J Inherit Metab Dis. 2019;42:655-72.
- [CrossRef] [PubMed] [Google Scholar]
- Aicardi-Goutieres syndrome: Neuroradiologic findings and follow-up. Am J Neuroradiol. 2009;30:1971-6.
- [CrossRef] [PubMed] [Google Scholar]
- Neuroimaging in cockayne syndrome. Am J Neuroradiol. 2010;31:1623-30.
- [CrossRef] [PubMed] [Google Scholar]
- Intracranial calcifications in childhood: Part 2. Pediatr Radiol. 2020;50:1448-75.
- [CrossRef] [Google Scholar]




