

Translate this page into:
Acrocallosal syndrome: Classical findings in a case report with a literature review
*Corresponding author: Siddhi Chawla, Department of Radiology, Sardar Patel Medical College, Bikaner, Rajasthan, India. siddhi.chawla870@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Chawla S, Charan A. Acrocallosal syndrome: Classical findings in a case report with a literature review. Case Rep Clin Radiol. doi: 10.25259/CRCR_143_2023
Abstract
We present a case of a 1-day-old neonate with classical findings of acrocallosal syndrome. The child presented with difficulty in feeding. Clinically, the patient had craniofacial anomalies and digital malformations. Imaging with ultrasonography and magnetic resonance imaging revealed characteristic features of corpus callosum agenesis with dandy walker malformation. A classical case of acrocallosal syndrome with sporadic occurrence is discussed with a detailed review of all such previously reported cases in the literature till date and a discussion of possible differentials.
Keywords
Acrocallosal syndrome
Agenesis of corpus callosum
Polydactyly
Magnetic resonance imaging
Pediatric radiology

INTRODUCTION
Acrocallosal syndrome (ACS) was first described by Schinzel in 1979,[1] as a conundrum of findings, including post-axial polydactyly, duplication of the hallux, and abnormality of corpus callosum including agenesis or dysgenesis, occasional anencephaly, and/or Dandy–Walker malformation, characteristic craniofacial abnormalities, and moderate-to-severe mental retardation. In clinical practice, however, the clinical, physical, and imaging findings vary over a spectrum and hence vary with the age of presentation. The inheritance pattern of ACS is autosomal recessive with the gene situated on chromosome 12p caused due to mutations in the KIF7 gene;[2,3] however, sporadic cases have also been reported.[4] Our case describes the classical findings in ACS in a 1-day-old neonate which is the second case presented at this age according to the reported literature.[4]
CASE PRESENTATION
A 1-day-old full-term male child delivered vaginally as a result of non-consanguineous marriage presented to the pediatric outpatient department (OPD) with a history of inability to suck and respiratory difficulty. He was the first-born child for the parents and no history of abortions was there. No antenatal scans were performed for the mother; however, the delivery was performed in a primary health center. The baby did not cry at birth and hence was referred to our hospital for management. The birth weight of the child was 2.7 kg (15–50th centile) and had a head circumference of 36 cm (85th centile). On physical examination, the baby had hyperteloric eyes with frontal bossing, a flat nasal bridge, and low-set ears [Figure 1]. There was pre-axial polydactyly in the left hand and post-axial polydactyly in the right hand, and both feet as well as duplication in both hallux with fusion of the right hallux [Figure 1]. Neurological examination showed mild hypotonia with sluggish reflexes. No history of abnormal body movements was present. Cardiovascular examination showed tachycardia with ejection systolic murmur. Abdominal and genitourinary examination was normal. Echocardiography revealed a patent foramen ovale with the normal position of the heart. Magnetic resonance imaging (MRI) brain revealed complete corpus callosum agenesis with large interhemispheric cysts not suppressed on fluid-attenuated inversion recovery sequences [Figure 2]. There was non-visualization of anterior falx with associated dysplasia of the left cerebral hemisphere with non-visualization of the frontal horn of the left lateral ventricle and atria of ventricle opening into a large cerebrospinal fluid intensity cyst in the interhemispheric region [Figure 3]. There was also associated bilateral cerebellar and vermian hypoplasia with a large posterior fossa cyst and torcular inversion consistent with Dandy–Walker malformation [Figures 2 and 3]. Ultrasound of the abdomen and pelvis was normal. Septic screen and metabolic profile were normal. The whole exome sequencing report revealed no particular gene mutation in our patient including the specific emphasis on the KIF7 gene. Parents were counseled regarding the sporadic nature of the disease in their child and the possible autosomal recessive nature of inheritance and were advised insertion of a cyst-peritoneal shunt by a neurosurgical team; however, they refused any surgical intervention and opted for treatment with home care and alternative sources of medicine. The patient since then was lost to follow-up.

- Photographs of patient show (a) hypertelorism and flat nasal bridge, (b) lateral profile shows low set ears, (c) hands show post-axial polydactyly in right hand and pre-axial polydactyly in left hand, and, (d) polysyndactyly in the right hallux and post axial polydactyly in left foot.
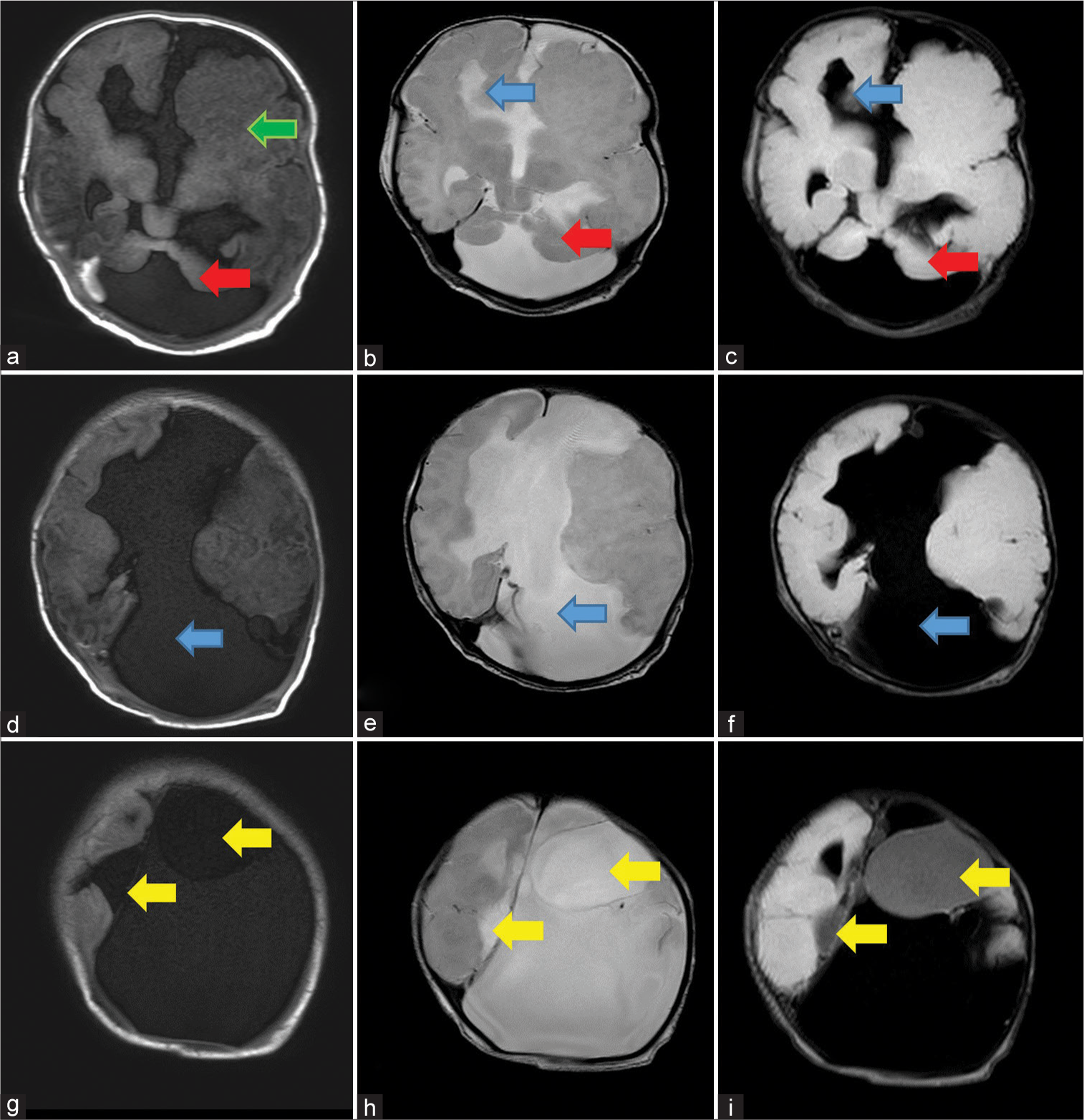
- Sequential axial sections of MRI shows: First row: (a) T1 weighted image shows dysmorphic left cerebral hemisphere with polymicrogyria (green arrow), bilateral cerebellar hypoplasia with a large posterior fossa cyst (red arrow), (b), T2 and (c) FLAIR weighted image, shows widely placed right frontal horn of lateral ventricle (blue arrows) and bilateral cerebellar hypoplasia with large posterior fossa cyst (red arrows), Second row: (d) T1 weighted image, (e) T2 weighted image and (f) FLAIR weighted images show non visualization of corpus callosum with large CSF intensity inter-hemispheric cyst (blue arrows seen in d-f), and Third row: (g) T1 weighted image, (h) T2 weighted image and (i) FLAIR weighted images shows few cysts which are not suppressing on FLAIR (yellow arrows seen in g-i).
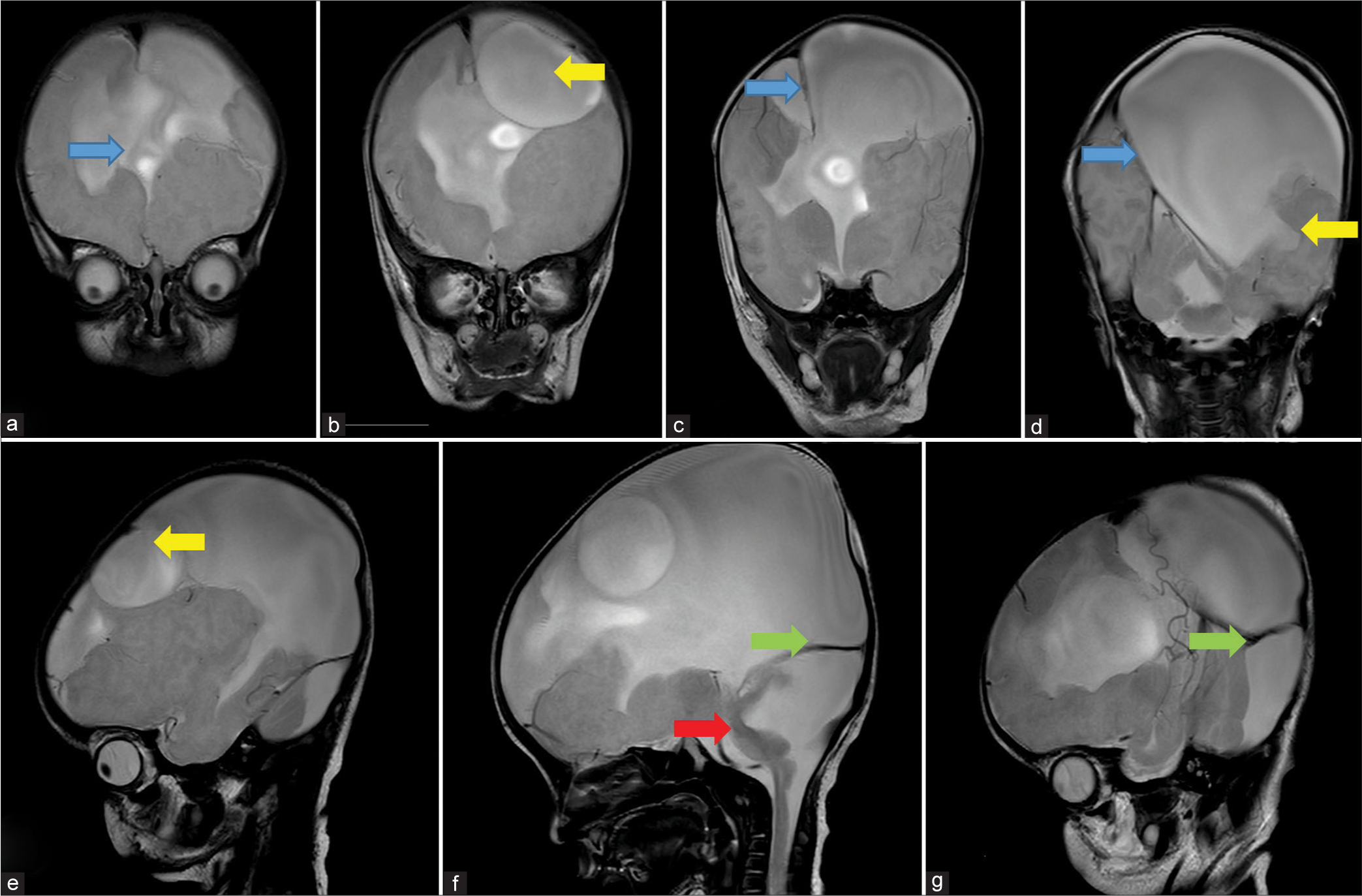
- Sequential coronal (a-d) and Sagittal (e-g) T2 weighted MRI images shows: (a) absence of anterior falx cerebri (blue arrow), (b) inter-hemispheric cyst (yellow arrow), (c) presence of falx cerebri in posterior aspect (blue arrow), (d) inter-hemispheric cyst communicating with lateral ventricle (yellow arrow) and presence of falx cerebri further posteriorly (blue arrow), (e) inter-hemispheric cyst (yellow arrow), (f) cerebellar hypoplasia with large posterior fossa cyst (red arrow) causing torcular inversion (green arrow) and, (g) High tentorium cerebelli signifying torcular inversion (green arrow).
DISCUSSION
ACS is rare with only a limited number of cases being described in the literature. To the best of our knowledge, this is the fifth reported case in the Indian population.[4,5] Most of the cases have reported an autosomal recessive inheritance and a history of consanguineous marriage.[6] Recent studies have shown mutations in the genes of kinesis KIF and transcription factor Gli3 which are involved in the ciliary signaling pathway involving the sonic hedgehog which is responsible for the formation of midline structures;[7] however, our case did not reveal any mutation in these genes, opening the possibility of an unknown gene mutation or sporadic occurrence. The disruptions in the development of the corpus callosum occur during the 5th–16th week of pregnancy.[5] Courtens et al. have given the diagnostic criteria for ACS as the presence of any three out of the four features, which include: (1) absence/hypoplasia/dysgenesis of the corpus callosum, (2) craniofacial dysmorphism, (3) polysyndactyly, and (4) psychomotor retardation.[8] Our case fulfilled all the given criteria in addition to having dandy walker malformation and cardiac involvement in the form of patent foramen ovale. With the increase in the number of cases being reported, there is a definite phenotypic variation as shown in [Table 1]. A variety of other syndromes are associated with agenesis of corpus callosum as well as polysyndactyly and due to overlap of phenotype, a differentiation of ACS from these syndromes is essential. [Table 2] summarizes the salient clinical features of these syndromes along with the differentiating features which help us to make a definite diagnosis of ACS. The prognosis of ACS depends on the degree of hypotonia and age of onset of seizures in addition to the associated malformations.[6] Life span of patients range from stillbirth to relatively normal lives with variable degrees of developmental delay.[5] A periodic follow-up of the patient with a pediatric neurologist, cardiologist, otolaryngologist, speech therapist, and child psychologist is recommended to the parents. Since there is a 25% chance of recurrence of this syndrome in the next child, genetic counseling of the parents is given utmost importance in detected cases.[5,6] Central nervous system malformations and polysyndactyly can be readily detected on antenatal scan and hence a judicious imaging follow-up in all future pregnancies is advised for mothers detected with ACS in one offspring.
| No. | Features | Authors | Our case |
|---|---|---|---|
| Clinical manifestations | |||
| 1. | Macrocephaly | Bhambal et al.(2015),[5] Subramanian et al.(2019),[7] Revanna et al. (2018),[9] Ibisler et al.(2015),[10] Schinzel and Schmid (1980)[11] | + |
| 2. | Seizures/abnormal EEG | Revanna et al. (2018),[9] Schinzel et al. (1986),[12] Koenig et al.(2002)[13] | − |
| 3. | Hypotonia | Bhambal et al.(2015),[7] Yuksel et al.(1990),[14] | + |
| 4. | Hypertelorism and frontal bossing | Bhambal et al.(2015),[5] Subramanian et al.(2019),[7] Ibisler et al.(2015),[10] Schinzel (1988),[15] Hosdurg et al.(2018),[16] | + |
| Neurological manifestations | |||
| 5. | Agenesis/hypoplasia of corpus callosum | Bhambal et al.(2015),[5] Lamrissi et al.(2022),[6] Subramanian et al.(2019),[7] Revanna et al. (2018),[9] Ibisler et al.(2015),[10] Hosdurg et al.(2018),[16] Moeachler et al.(1987)[17] | + |
| 6. | Cerebellar hypoplasia (including dandy walker malformation) | Ibisler et al.(2015),[10] Hosdurg et al.(2018),[16] Hendrik et al.(1990)[18] | + |
| 7. | Brainstem dysplasia, joubert syndrome, hippocampal malrotation | Subramanian et al.(2019)[7] | − |
| 8. | Optic atrophy | Hendriks et al. (1990)[18] | − |
| 9. | Olfactory bulb agenesis and olfactory tract abnormalities | Subramanian et al.(2019),[7] | − |
| Musculoskeletal manifestations | |||
| 10. | Hallux duplication | Ibisler et al.(2015),[10] Schinzel and Schmid (1980),[11] Hosdurg et al.(2018)[16] | + |
| 11. | Pre-and post-axial polydactyly of toes and fingers | Revanna et al. (2018),[9] Ibisler et al.(2015),[10] Schinzel (1988),[15] Hendriks et al.(1990)[18] | + |
| 12. | Cranial synostosis | Subramanian et al.(2019)[7] | − |
| 13. | Orofacial malformations | Bhambal et al.(2015),[5] Lamrissi et al.(2022),[6] Hosdurg et al.(2018)[16] | − |
| 14. | Rib anomalies | Singhal et al.(2014)[4] | − |
| 15. | CTEV | Hosdurg et al.(2018)[16] | − |
| Cardiac manifestations | |||
| 16. | Congenital heart disease | Ibisler et al. (2015),[10] Hosdurg et al. (2018),[16] Casamassima et al.(1989)[19] | + |
| 17. | Dextro position of the heart | Singhal et al.(2014)[4] | − |
| 18. | Cyanotic spells | Yuksel et al. (1990)[14] | − |
| Others | |||
| 19. | Delayed fall of cord | Singhal et al. (2014)[4] | + |
CTEV: Congenital talipus equinus varus, EEG: Electroecephalogram
| Classical features | Differentiating features | |
|---|---|---|
| Syndromes associated with corpus callosum agenesis | ||
| Aicardi syndrome | Infantile spasms, chorioretinal lacunae, microcephaly, polymicrogyria, porencephalic cysts; enlarged cerebral ventricles due to hydrocephalus | No association of polysyndactyly; Absence of seizures |
| Andermann syndrome | Motor and sensory neuropathy, mild mental retardation, mild facial dysmorphism (including hypertelorism, short nose, broad nasal root, high arched palate, elongated facies, and ptosis) | No association of polysyndactyly, Absence of neuropathy |
| Shapiro syndrome | Paroxysmal hypothermia, hyperhidrosis | Absence of clinical features, absence of polysyndactyly |
| DiGeorge syndrome | Congenital heart disease, defects in the palate, neuromuscular problems, learning disabilities, mild differences in facial features, and recurrent infections | Absence of polysyndactyly, absence of hypotonia |
| Syndromes associated with polysyndactyly | ||
| Greig cephalopolysyndactyly syndrome | Hypertelorism, macrocephaly, frontal bossing, and polysyndactyly | Absence of central nervous system anomalies |
| Oro-facial-digital syndrome type II | Hypertelorism, polysyndactyly, cleft lip and palate, conductive deafness, choroidal coloboma, renal and cardiac defects | Absence of hypotonia, Absence of central nervous system anomalies, and Absence of clinical features |
| Smith-Lemli-Opitz syndrome (7-dehydroxycholesterol reductase deficiency) | Psychomotor retardation, microcephaly, hypotonia, holoprosencephaly, low-set ears, micrognathia, polysyndactyly, cardiac anomalies, and urogenital anomaly | Absence of classical corpus callosum anomalies, Absence of macrocephaly |
| Rubenstein Taybi syndrome | Facial dysmorphism, limb anomalies, microcephaly, intellectual retardation, congenital cardiac abnormalities, and hirsutism | Absence of classical corpus callosum anomalies, Absence of macrocephaly, and absence of psychomotor retardation |
CONCLUSION
ACS is a rare anomaly and only a few cases have been reported in the Indian population. Imaging plays an important role in antenatal diagnosis of this condition. Postnatally, imaging is essential for the diagnosis of corpus callosum agenesis along with other associated cranial, facial, and digital anomalies that distinguish it from other syndromes. Early recognition of the condition is necessary to intervene surgically and to counsel the parents regarding the genetics of the disease and dedicated follow-up of the affected child in special OPDs. Molecular testing should be done in all suspected cases.
TEACHING POINTS
ACS is rare and a conundrum of clinical features such as facial and digital anomalies (polydactyly) should prompt the clinician to perform a radiological investigation with an MRI brain to look for corpus callosum anomalies.
Genetic testing is suggested for all cases with suspected ACS to look for the KIF7 gene and yet unknown mutations that can present with similar morphology.
MCQs
-
Which of the following clinical features is not a criterion to diagnose acrocallosal syndrome?
Psychomotor retardation
Polydactyly
Seizures
Craniofacial dysmorphism
Answer Key: c
-
Which are the essential MRI findings to diagnose acrocallosal syndrome?
Corpus callosum dysgenesis
Dandy walker malformation
Schizencephaly
Hydrocephalus
Answer Key: a
-
Which of the following is not associated with microcephaly?
Aicardi syndrome
Acrocallosal syndrome
TORCH infection
Apert syndrome
Answer Key: b
Ethical approval
The research was in compliance with Helsinki declaration 1964.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Postaxial polydactyly, hallux duplication, absence of the corpus callosum, macrencephaly and severe mental retardation: A new syndrome? Helv Paediatr Acta. 1979;34:141-6.
- [Google Scholar]
- Acrocallosal syndrome in a child with de novo inverted tandem duplication of 12p11.2-p13.3. Ann Genet. 1992;35:41-6.
- [Google Scholar]
- KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet. 2011;43:601-6.
- [CrossRef] [PubMed] [Google Scholar]
- The acrocallosal syndrome in a neonate with further widening of phenotypic expression. Iran J Child Neurol. 2014;8:60-4.
- [Google Scholar]
- Orodental manifestations in cases with partial agenesis of corpus callosum-rare phenomena. J Oral Biol Craniofac Res. 2015;5:106-11.
- [CrossRef] [PubMed] [Google Scholar]
- Acro-callous syndrome: A case report. Int J Surg Case Rep. 2022;96:107210.
- [CrossRef] [PubMed] [Google Scholar]
- Olfactory bulb and olfactory tract abnormalities in acrocallosal syndrome and Greig cephalopolysyndactyly syndrome. Pediatr Radiol. 2019;49:1368-73.
- [CrossRef] [PubMed] [Google Scholar]
- Acrocallosal syndrome in an Algerian boy born to consanguineous parents: Review of the literature and further delineation of the syndrome. Am J Med Genet. 1997;69:17-22.
- [CrossRef] [Google Scholar]
- Agenesis of the corpus callosum with interhemispheric cyst: Clinical implications and outcome. BMJ Case Rep. 2018;11:bcr2018227366.
- [CrossRef] [PubMed] [Google Scholar]
- Novel KIF7 mutation in a Tunisian boy with acrocallosal syndrome: Case report and review of the literature. Mol Syndromol. 2015;6:173-80.
- [CrossRef] [PubMed] [Google Scholar]
- Hallux duplication, postaxial Polydactyly, absence of corpus callosum, severe mental retardation and additional anomalies in two unrelated patients: A new syndrome. Am J Med Genet. 1980;6:241-9.
- [CrossRef] [PubMed] [Google Scholar]
- The acrocallosal syndrome in sisters. Clin Genet. 1986;30:399-405.
- [CrossRef] [PubMed] [Google Scholar]
- Spectrum of acrocallosal syndrome. Am J Med Genet. 2002;108:7-11.
- [CrossRef] [PubMed] [Google Scholar]
- The acrocallosal syndrome in a Turkish Boy. J Med Genet. 1990;27:48-9.
- [CrossRef] [PubMed] [Google Scholar]
- The acrocallosal syndrome in first cousins: Widening of the spectrum of clinical features and further support for autosomal inheritance. J Med Genet. 1988;25:332-6.
- [CrossRef] [PubMed] [Google Scholar]
- Anaesthetising an infant with acrocallosal syndrome: An unusual case. Indian J Anaesth. 2018;62:389-91.
- [CrossRef] [PubMed] [Google Scholar]
- Acrocallosal syndrome: New findings. Am J Med Genet. 1989;32:195-199.
- [CrossRef] [PubMed] [Google Scholar]
- Acrocallosal Syndrome: Additional manifestation. Am J Med Genet. 1989;32:311-7.
- [CrossRef] [PubMed] [Google Scholar]




