

Translate this page into:
Clinico-radiological correlation in a case of Schuurs–Hoeijmakers syndrome
*Corresponding author: Swaksh Pradeep Nemani, Department of Radio-Diagnosis, Seth G S Medical College and KEM Hospital, Mumbai, Maharashtra, India. dr.swaksh113@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Bolke P, Nemani SP, Mettan O. Clinicoradiological correlation in a case of Schuurs–Hoeijmakers syndrome. Case Rep Clin Radiol. doi: 10.25259/CRCR_88_2024
Abstract
Schuurs–Hoeijmakers syndrome is a very rare disease entity presenting as a neuro-developmental disorder with dysmorphic facies and a host of non-specific phenotypes. The diagnosis of this autosomal dominant disorder is made by genetic testing based on high clinical suspicion. Little is known about the radiological findings in such children. Here, we show a case of a 7-year-old boy diagnosed by whole exome sequencing and portray the various radiological and clinical findings that he presented with.
Keywords
Schuurs–Hoeijmakers syndrome
Facial dysmorphism
Neurodevelopmental disorder

INTRODUCTION
Schuurs–Hoeijmakers syndrome is a very rare disease entity presenting as a neuro-developmental disorder with dysmorphic facies and a host of non-specific phenotypes. The diagnosis of this autosomal dominant disorder is made by genetic testing based on high clinical suspicion. Little is known about the radiological findings in such children. Here, we show a case of a 7-year-old boy diagnosed with whole exome sequencing and portray the various radiological and clinical findings that he presented.
CASE REPORT
A 7-year-old boy, third by birth order, born to a non-consanguineous married couple presented with global developmental delay and dysmorphic facies. He was delivered by lower-segment cesarean section due to maternal factors and had history of neonatal intensive care unit (ICU) stay for 7 days in view of neonatal hypotonia and respiratory distress. He had clinical indications of bilateral mild hearing loss with brainstem evoked response audiometry (BERA) showing mild bilateral sensorineural hearing loss. He had nil language output with minimal comprehension of verbal speech and had not been schooled. He had history of recurrent hospital admissions for lower respiratory tract infection (LRTI) not warranting ICU stay. He also had history of 4 episodes of unprovoked generalized tonic-clonic seizures since the age of 5 years. There was no similar history of psycho-motor retardation or seizure disorder in his siblings or parents.
On physical examination, he had bilateral low set ears, hypertelorism, flattened philtrum, down-slanting eyes, ptosis, bulbous nose, thin lips, widely-spaced teeth, and a flattened occiput [Figure 1]. He was subjected to an erect chest radiograph which revealed cardiomegaly with upturned apex and pulmonary plethora suggestive of congenital heart disease [Figure 2]. The radiography report coupled with history of recurrent LRTIs prompted a 2D ECHO which revealed an atrial septal defect, ventricular septal defect, and patent ductus arteriosus with the left to right shunt [Figure 2]. On the basis of male gender, congenital heart defect and dysmorphic facies, Noonan Syndrome was suspected and a whole exome sequencing was done which revealed c.607C >T (p.Arg203Trp) heterozygous pathogenic variant of phosphurin acidic cluster sorting protein 1 (PACS 1) gene suggestive of Schuurs–Hoeijmakers syndrome.
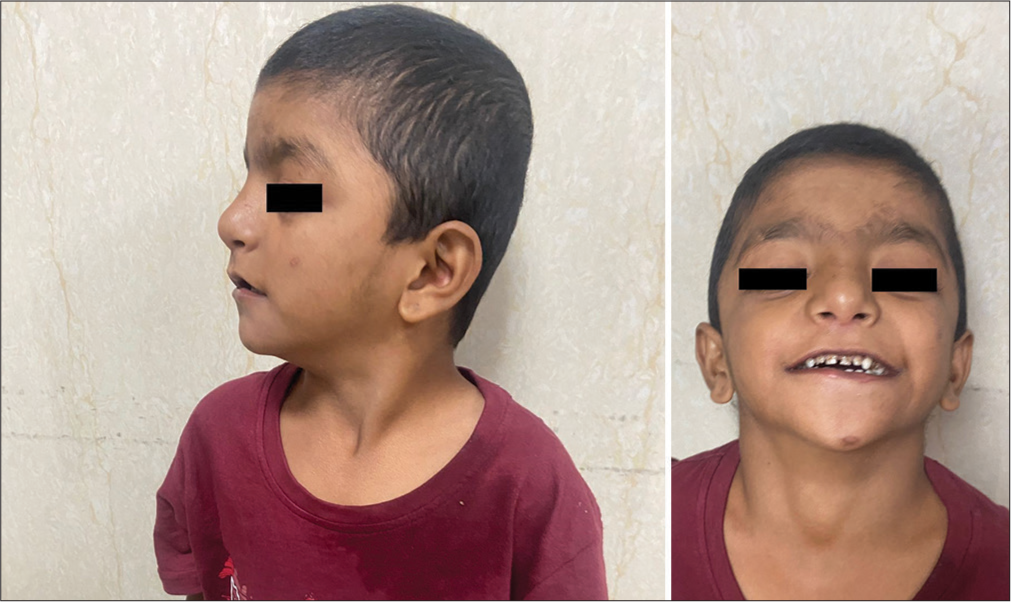
- (a) Profile clinical image shows low-set ears and flat occiput. (b) Frontal clinical image shows hypertelorism, widely-spaced teeth, bulbous nose, flattened philtrum, and thin lips.
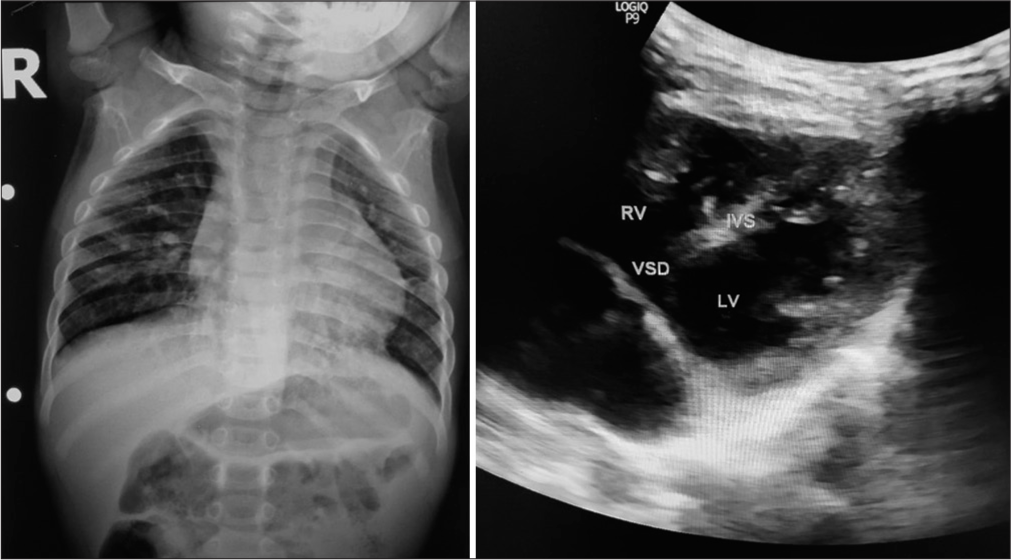
- Frontal chest radiograph shows cardiomegaly with upturned apex and pulmonary plethora suggestive of congenital heart disease. 2D ultrasound image using curvilinear probe at 9 Hz in left 5th intercostal space in a four-vessel view shows the high ventricular septal defect. RV = Right Ventricle; LV = Left Ventricle; VSD = Ventricular Septal Defect; IVS = Inter Ventricular Septum.
An magnetic resonance imaging (MRI) brain was done to evaluate for possible structural malformations being the etiology of the seizures. Multi-planar, multi-echo MRI brain was performed on a 1.5T Siemens console. Findings observed included
-
Dilatation of the occipital horns of lateral ventricles – colpocephaly [Figure 3]
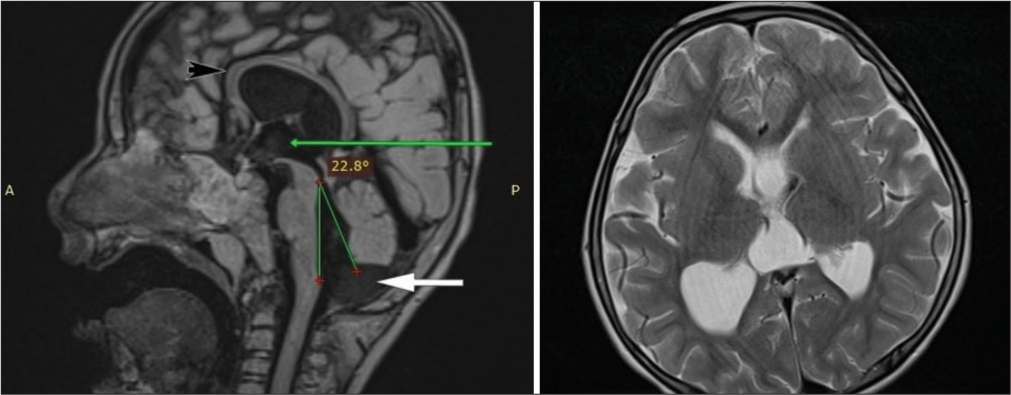 Figure 3:
Figure 3:- T1 Magnetization-Prepared Rapid Acquisition Gradient Echo (MPRAGE) mid-sagittal image showing inferior vermian hypoplasia (white arrow), with tegmento-vermian angle of 22°, dilated third ventricle (green arrow) and thinned-out corpus callosum throughout its substance (black arrow) with upward bowing. T2 axial image showing dilatation of bilateral occipital horns of lateral ventricles suggestive of colpocephaly.
Thinned out corpus callosum throughout its course suggesting corpus callosal dysgenesis [Figure 3]
Asymmetric dilatation of both lateral ventricles, third and fourth ventricle with upward bowing of the thin corpus callosum suggesting compensated communicating hydrocephalus
Inferior vermian hypoplasia with a tegmento-vermian angle of 22° [Figure 3]
Persistent cavum septum vergae.
The child was advised expectant management for the congenital heart disease and prescribed anti-epileptics for the seizure episodes with routine outpatient department basis follow-up.
DISCUSSION
Schuurs–Hoeijmakers syndrome is a very rare disease entity with only 61 published cases worldwide as of May 2021 as per our literature search.[1] The syndrome, also known as PACS1 neurodevelopmental disorder (NDD), is autosomal dominant by inheritance but is caused in most cases by a de novo missense mutation in the PACS1 gene on chromosome 11q13.1.[2] PACS1 is a membrane traffic regulator which facilitates transport of proteins from the Golgi complex to the endosomes.[3] It plays an essential role in cellular trafficking of numerous cytosolic proteins and its absence can cause varied multi-systemic involvement. PACS1 mRNA expression is maximal during the period of fetal neurogenesis probably accounting for the predominant craniofacial and neurological deformities in children with Schuurs– Hoeijmakers syndrome.[4] Despite being an autosomal dominant disorder, there are de novo mutations in parental gonads or the developing fetus accounting for siblings and parents of the index case not having corresponding phenotypic features.[5]
Schuurs–Hoeijmakers syndrome is predominantly a NDD with dysmorphic facies. Tenorio-Castaño et al., in 2021, performed a literature review of all previously documented cases and enlisted the most frequent symptoms and presenting features of this syndrome in order of prevalence.[1] They found that the most common presenting symptoms were intellectual disability, dysmorphic facies, speech delay, and seizures, all of which were present in our patient. Facial features commonly encountered were bulbous nose, microcephaly, hypertelorism, abnormal skull shape, thin upper lip, and low set ears in decreasing order of prevalence. However, none of these were found to be specific for Schuurs–Hoeijmakers syndrome. Rare systemic involvements included gastro-esophageal reflux and congenital heart disease. One case report from Iraq in 2021 mentioned MRI brain findings including mild cerebral atrophy, bilateral retinal colobomas, and thinning of the corpus callosum.[6] However, our literature search did not yield other studies in which neuroimaging findings were elaborated.
Thus, in view of lack of pathognomonic features, high index of suspicion coupled with genetic testing is mandatory for diagnosis of this rare syndrome. The differential diagnosis includes a host of genetic disorders manifesting as facial dysmorphism with neurodevelopmental delay. Initial molecular testing includes a chromosomal microarray analysis. If a definitive diagnosis is not reached, whole exome sequencing or multigene panel can be employed.[7] Little is known regarding the life expectancy and prognosis of this disorder.
DIFFERENTIAL DIAGNOSIS
Imaging differential diagnosis based on MRI findings includes
Posterior fossa malformations of the Dandy–Walker spectrum
Trisomy 18
PHACES syndrome (posterior fossa malformations, congenital heart defect).
CONCLUSION
Schuurs–Hoeijmakers syndrome is a rare NDD with not many documented cases worldwide. It has no classical clinical features; hence, diagnosis relies heavily on genetic testing. Our case helps expand the existing knowledge on presenting features and elaborates neuro-structural anomalies on MRI.
TEACHING POINTS
In patients with dysmorphic facies and intellectual disability, it is essential to suspect neuro-structural anomalies and perform an MRI brain.
In patients with vermian hypoplasia, look for associated supra-tentorial anomalies like corpus callosal dysgenesis.
MCQs
-
Colpocephaly is dilatation of which horn of the lateral ventricle?
Frontal
Occipital
Temporal
Body
Answer Key: b (Occipital)
-
Tegmento-vermian angle in Vermian Hypoplasia is?
0°
<18°
18–45°
>45°
Answer Key: c (18–45°)
-
Which of the following is not seen in Schuurs Hoiejmakers Syndrome?
Persistent cavum septum vergae
Vermian hypoplasia
Corpus callosal dysgenesis
Macrocephaly
Answer Key: d (Macrocephaly)
Acknowledgement
We would like to acknowledge the relatives of our patient for their informed consent to present clinical images while maintaining anonymity.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Schuurs-Hoeijmakers syndrome (PACS1 neurodevelopmental disorder): Seven novel patients and a review. Genes. 2021;12:738.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical delineation of thePACS1-related syndrome-Report on 19 patients. Am J Med Genet Part A. 2016;170:670-5.
- [CrossRef] [PubMed] [Google Scholar]
- PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell. 1998;94:205-16.
- [CrossRef] [PubMed] [Google Scholar]
- Gene expression across mammalian organ development. Nature. 2019;571:505-9.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular basis of the Schuurs-Hoeijmakers syndrome: What we know about the gene and the PACS-1 protein and novel therapeutic approaches. Int J Mol Sci. 2022;23:9649.
- [CrossRef] [PubMed] [Google Scholar]
- Schuurs-Hoeijmakers syndrome in a patient from Iraq-Kirkuk. Clin Case Rep. 2021;9:e04897.
- [CrossRef] [PubMed] [Google Scholar]
- PACS1 neurodevelopmental disorder. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 1993-2024.
- [Google Scholar]




