

Translate this page into:
Multimodality approach in tetraphocomelia – An uncommonly seen red flag in anomaly scan

*Corresponding author: Jyoti Gupta, Department of Radiodiagnosis and Interventional Radiology, Vardhman Mahavir Medical College and Safdarjung Hospital, New Delhi, India. jyotigupta99@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Nigam K, Gupta J, Chawla N. Multimodality approach in tetraphocomelia – An uncommonly seen red flag in anomaly scan. Case Rep Clin Radiol. doi: 10.25259/CRCR_16_2024
Abstract
Robert’s syndrome is an autosomal recessive genetic disorder characterized by fetal growth restriction along with a myriad of limb and craniofacial abnormalities such as tetraphocomelia, oligodactyly, clinodactyly, midline facial clefts, midfacial masses, hypertelorism, and ear malformations. Spinal anomalies have rarely been reported in previous literature. Herein, we report one such rare proven case of Robert’s syndrome, diagnosed antenatally and managed by medical termination of pregnancy.
Keywords
Robert’s syndrome
Fetal medicine
Antenatal anomaly scan
Fetal ultrasound
Fetal magnetic resonance imaging
Establishment of cohesion 1 homologue 2 spectrum disorder

INTRODUCTION
Roberts syndrome is an extremely rare cause of tretraphocomelia and is a cytogenetic establishment of cohesion 1 homologue 2 (ESCO 2) spectrum disorder of autosomal recessive inheritance; characterized by limb deformities, craniofacial abnormalities and fetal growth restriction.[1] The inciting factor is mutation in ESCO2 gene located on chromosome 8(2); with resultant loss of acetyltransferase enzyme activity; leading to premature centromere separation in metaphase chromosomes.[1] Detection of this syndrome in early antenatal period alerts physicians for better post natal care or medical termination of pregnancy if required; to save undue physical and mental burden on the mother and family; owing to higher mortality rates in fetuss and newborns with equally high morbidity in surviving individuals. We report one such antenatally diagnosed cytogenetically proven case highlighting the role of multimodality imaging in such cases.
CASE REPORT
A 32-year-old pregnant female patient came to the Radiology Department for a level II anomaly scan. This was the second conception in a non-consanguineous marriage with an insignificant maternal history. No previous ultrasound was done. A single live intrauterine fetus with symmetrical fetal growth restriction in cephalic presentation was seen, with an average gestational age of 18 weeks and 2 days by ultrasonography (USG), and an array of other anomalies, the most striking being thetetraphocomelia. In upper limbs, two parallelly arranged hypoplastic bones were seen, likely the forearm bones, with no visualization of bilateral humeri [Figure 1a]. Both hands showed only three fingers suggestive of oligodactyly, as was also seen on 3D ultrasound [Figure 1b]. Bilateral lower limbs showed non-parallel orientation with syndactyly [Figure 1c]. The fetal face revealed hyperteleorism with midline facial clefts, absent nasal bones, and normal fetal ears [Figure 1c-d]. A hypoechoic lobulated soft tissue without any Doppler perceptive flow was seen in the midline facial clefts protruding through a possible anterior cranial fossa floor defect. Central nervous system evaluation showed frontal bossing with a disproportionately increased occipitofrontal diameter and a relatively small posterior fossa. Enlargement of bilateral lateral ventricles with colpocephaly and asymmetry of lateral ventricles was seen [Figure 1e and 1f]. There was no visualization of the rostrum and body of the corpus callosum with normal optic chiasma, suspicious of corpus callosal dysgenesis. On Doppler evaluation, paired anterior cerebral arteries were visualized. On fetal spine evaluation, persistent cervical neck hyperextension, loss of sacral uptilt, and no visualization of lower lumbar and sacral vertebrae with closely apposed iliac bones were seen as suggestive of lumbosacral dysgenesis [Figure 2a and b]. There was situs solitus with levocardia, levoposition, and a small thoracic circumference compared to the abdominal circumference with an increased cardiothoracic ratio (>0.6).
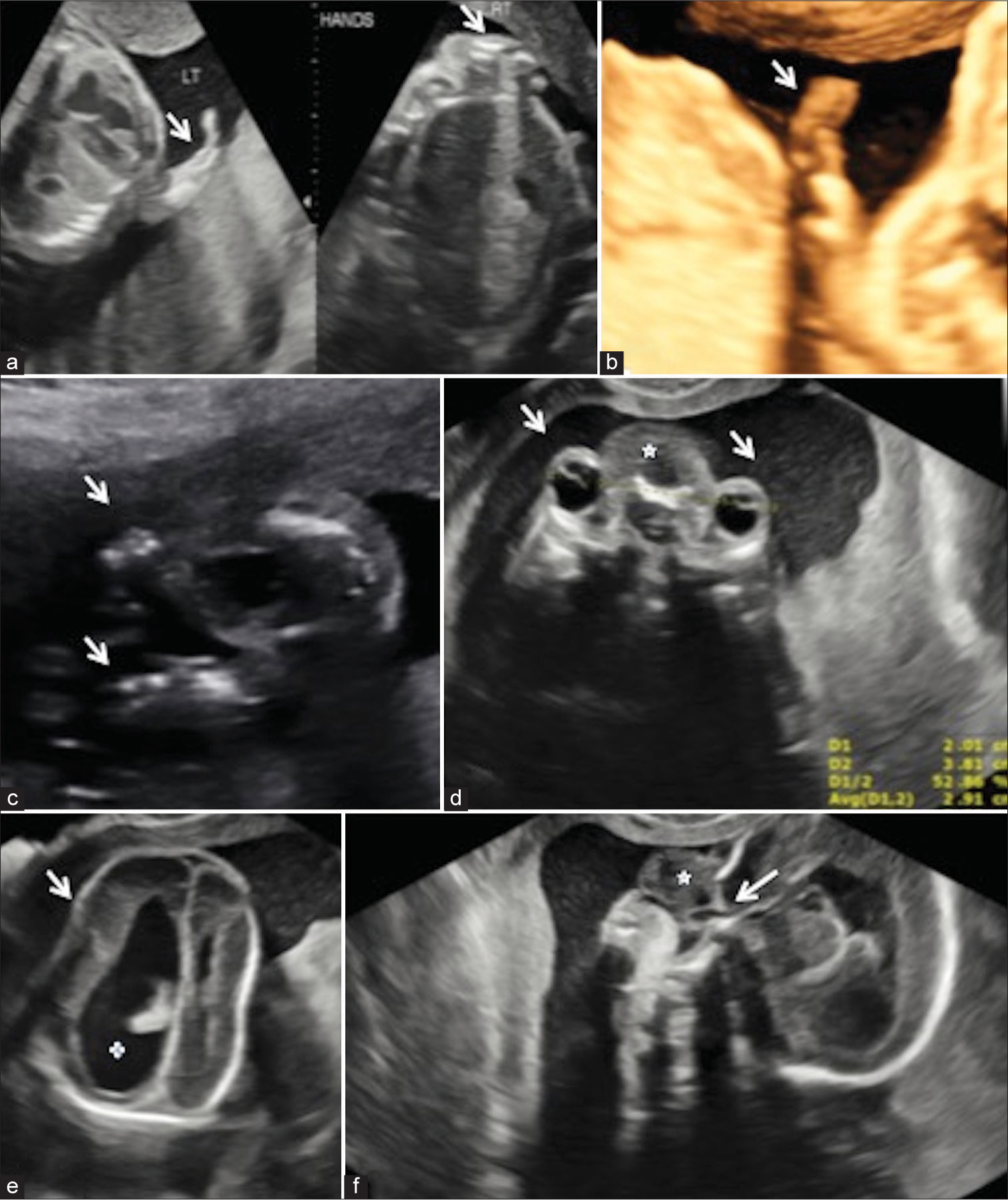
- (a) Bilateral upper limbs (white arrows) appear severely shortened with absence of bilateral humeri. Two parallelly arranged hypoplastic bones (labelled right) can be seen in right limb. (b) 3D ultrasonography (USG) image shows a small upper limb with direct attachment with the thorax. Three digits (white arrow) can be appreciated in the hand suggestive of oligodactyly. (c) Bilateral lower limbs show severe shortening with direct attachment (white arrows) to the fetal body. (d) Axial ultrasound Section through orbits showed wide apart eyes (white arrows) with increased inner orbital distance. A hetergeneous hypoechoic lesion (white star) is seen in the interorbital region. (e) Axial image of the skul shows mild frontal bossing (white arrow) with asymmetrically dilated lateral ventricles and colpocephaly (white cross). (f) Sagittal USG images of face showed a heterogeneously hypoechoic structure (white star) protruding through the midline facial defect (white arrow).
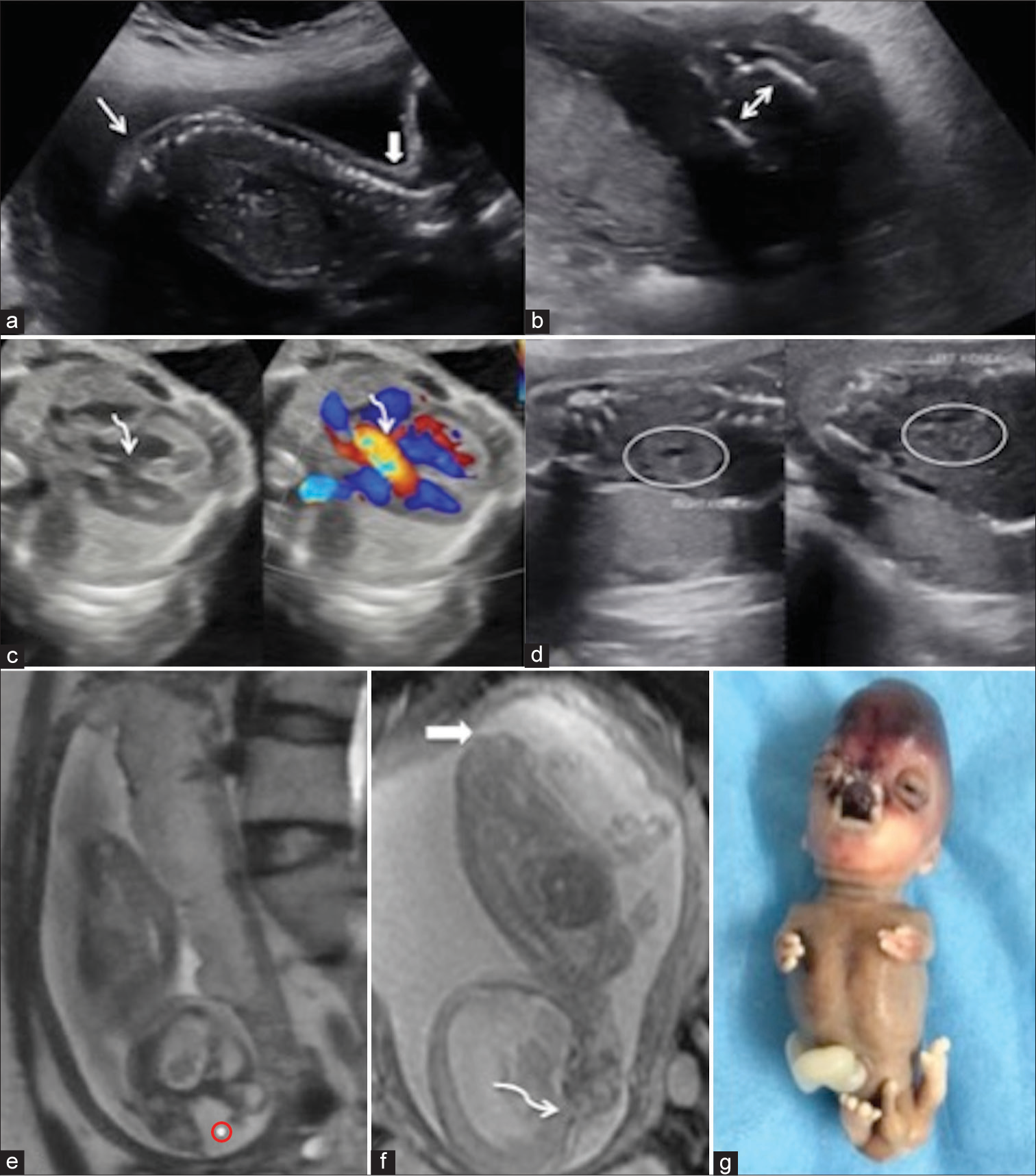
- (a) Sagittal image of Fetal spine showed loss of sacral uptilt (white arrows) with no visualization of sacral and lower lumbar vertebrae. (b) Axial section through the pelvis showed mildly apposed iliac bones (double sided white arrow) suggesting lumbosacral dysgenesis. (c) Gray scale axial ultrasonography image through chest shows a 4 chambered heart with a defect in interventricular septum in perimembranous region (curved white arrow) showing color doppler flow (curved white arrow). (d) Bilateral kidneys (white oval circles) appear enlarged (right more than left) and show few anechoic cysts within. The renal parenchyma in right kidney also shows raised echogenicity. (e) Fetal magnetic resonance imaging (MRI) shows midline defect in nasoethmoidal region with protrusion of a T2 hyperintense mass (red circle with dot) in midline. (f) Sagittal MRI images through face show continuity of the tissue with cerebral parenchyma (curved white arrow) with Slight T2 heterointense lesion in the sacral region (solid white arrow). (g) Post-medical termination of pregnancy clinical image of fetus showing tetraphocomelia with tetraoligodactyly, dysmorphic facial morphology with frontal bossing, midline facial cleft, hypertelorism, dysplastic cerebral tissue in nasoethmoidal region showed blackish appearance. Enlarged phallus could be appreciated.
Atrioventricular and ventriculoarterial concordance was seen with a tiny echogenic intracardiac focus in the left ventricle. A small perimembranous ventricular septal defect with aorto-septal discontinuity was seen on the gray scale USG and color Doppler [Figure 2c]. Abdominal examination showed bilateral renomegaly with a hypoechoic halo and a few bilateral small anechoic cysts [Figure 2d]; suggesting the possibility of bilateral cystic renal dysplasia. The rest of the fetal examination was unremarkable.
A tailored fetal magnetic resonance imaging (MRI) scan depicted a T2 hyperintense mass through the midline defect, showing continuity with the cerebral parenchyma of the frontal lobes, likely representing dysplastic cerebral tissue [Figure 2e]. Fetal MRI also confirmed tetrapholomelia with bilateral upper limbs showing three digits, persistent cervical hyperextension, partial corpus callosal agenesis, and midline facial clefts. Low-lying spinal cord with the presence of a skin-covered heterogeneously hypointense T2-weighted subcutaneous mass in the sacral region suggests a closed type of spina dysraphism [Figure 2f], which was not well appreciated on the USG exam.
Based on these constellations of findings with tetraphocomelia, a possibility of syndromic association, likely Robert’s syndrome, was kept. The patient underwent medical termination of pregnancy and whole exome gene sequencing, which revealed Establishment of Cohesion 1 homologue 2 (ESCO 2): c.955+2_955+5delTAAG, confirming the diagnosis. The fetal specimen was sent for autopsy [Figure 2g], which revealed tetraphocomelia, oligodactyly with three digits in each hand and four digits in each foot, hypertelorism, a black-colored mass protruding in the midline face, and an enlarged phallus.
DISCUSSION
Robert’s syndrome, also known as hypomelia-hypotrichosis-facial hemangioma syndrome or Appelt-Gerken-Lenz syndrome, was first described by John Roberts in 1919. It is an extremely rare cytogenetic ESCO2 spectrum disorder of autosomal recessive inheritance, characterized by limb deformities, craniofacial abnormalities, and fetal growth restriction.[1] The inciting factor is a mutation in the ESCO2 gene located on chromosome 8,[2] with a resultant loss of acetyltransferase enzyme activity, leading to premature centromere separation in metaphase chromosomes.[1] It is also known as pseudo-thalidomide syndrome since the structural abnormalities seen in Roberts also resemble the fetal abnormalities seen with a history of maternal thalidomide intake during pregnancy. The final diagnosis of Roberts syndrome is confirmed through cytogenetic analysis, which shows mutations in the ESCO2 gene. The clinical criteria for the diagnosis of Robert’s syndrome, first established by Vega et al. included growth retardation, symmetric mesomelic shortening of the limbs, and characteristic facies with microcephaly.[3] Craniofacial abnormalities in Roberts syndrome include hypertelorism, exophthalmos, downslanting palpebral fissures, cleft lip and cleft palate, brachydactyly, brachycephaly, midfacial hemangiomas, hypoplastic nasal alae, malar hypoplasia, micrognathia, corneal opacities, and ear malformations.[3] Limb abnormalities include phocomelia, brachydactyly, clinodactyly, arm-bone synostosis, club foot, and variable degrees of hypoplasia of the long bones of the upper and lower limbs. Other abnormalities include an enlarged phallus or clitoris, cryptorchidism, and cardiac defects. The majority of these defects were present in our case which helped in reaching a diagnosis with other ESCO spectrum disorders and thalidomide embryopathy as our differential diagnosis [Table 1].[1,4] Our case also showed the presence of spinal abnormalities which have not been reported till now. Genetic counseling of the parents is of paramount importance in such cases as the risk of recurrence in subsequent pregnancy is quite high, close to approximately 25%[4], as parents are obligate heterozygotes of the mutant allele and siblings have a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected. It is usually associated with an adverse prognosis and high neonatal or childhood period mortality owing to cardiac and renal causes. Spinal dysraphism as seen in our case has seldom been reported in Roberts syndrome in previous literature, which may warrant further work-up for genetic associations.
| Syndrome | Features |
|---|---|
| Juberg- Haywards Syndrome | Microcephaly, upper limb hypoplasia and shortening, cleft lip/palate, short stature, delayed bone age and fifth digit clinodactyly. |
| Thrombocytopenia- Absent Radius | Bilateral absent radius with thrombocytopenia, cleft lip, cleft palate, limb, gastrointestinal and cardiovascular abnormalities. |
| Zimmer Tetraphocomelia | Facial clefts, anal atresia absent frontal bones, pulmonary hypoplasia, dysplastic kidneys, and genitourinary malformations. |
| Thalidomide embryopathy | Growth retardation; symmetric mesomelic shortening of the limbs and characteristic facies with microcephaly. |
CONCLUSION
Antenatal multimodality imaging including advanced techniques like 3D Ultrasound and fetal MRI provides an excellent opportunity for timely diagnosis of Roberts syndrome depicting the multisystemic structural abnormalities so that the informed choice for termination of pregnancy can be offered to the mother along with proper genetic counseling for future family planning.
TEACHING POINTS
Tetraphocomelia in a fetus should raise the suspicion of thalidomide and ESCO gene spectrum disorders warranting a further detailed evaluation using 3D Ultrasound and fetal MRI.
Proper genetic counseling for future family planning should be advised to the couple.
MCQs
-
Tetraphocomelia is seen in
Robert’s syndrome.
ESCO gene spectrum disorders.
Thalidomide embryopathy.
All of the above.
Answer Key: d
-
ESCO 2 gene spectrum disorders are all except-
Robert’s syndrome
Juberg-Haywards Syndrome
Thrombocytopenia- Absent Radius
SC- phocomelia
Answer Key: c
-
The facial anomalies commonly seen in Robert’s syndrome are all except-
Cleft lip
Cleft palate
Facial hemangioma
Facial dermoid cyst.
Answer Key: d
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Roberts syndrome In: Obstetric imaging: Fetal Diagnosis and Care. Netherlands: Elsevier; 2018. p. :577-8.
- [CrossRef] [Google Scholar]
- A novel frameshift mutation in ESCO2 gene in Roberts syndrome. J Coll Physicians Surg Pak. 2018;28:403-5.
- [CrossRef] [PubMed] [Google Scholar]
- Phenotypic variability in 49 cases of ESCO2 mutations, including novel missense and codon deletion in the acetyltransferase domain, correlates with ESCO2 expression and establishes the clinical criteria for Roberts syndrome. J Med Genet. 2010;47:30-7.
- [CrossRef] [PubMed] [Google Scholar]
- GeneReviews® ESCO2 spectrum disorder Seattle, WA: University of Washington, Seattle; 1993-2023.
- [Google Scholar]




