

Translate this page into:
A bizarre case of recurrent hemoptysis and pneumothorax in a young male

*Corresponding author: Tharun Shaju, Department of Radiodiagnosis, Pushpagiri Institute of Medical Sciences and Research Centre, Pathanamthitta, Kerala, India. tharunshaju7@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Shaju T, Benjamin G, Mangalanandan S, Johncy KJ. A bizarre case of recurrent hemoptysis and pneumothorax in a young male. Case Rep Clin Radiol. doi: 10.25259/CRCR_77_2024
Abstract
Vascular Ehlers–Danlos syndrome (vEDS) is a rare inherited disorder affecting approximately 1 in 150,000 individuals, caused by heterozygous mutations in the COL3A1 gene. Characterized by primary spontaneous pneumothorax and severe connective tissue fragility, vEDS remains poorly understood clinically, making diagnosis challenging. We present the case of a 20-year-old male with a history of mitral valve prolapse, recurrent hemoptysis, knee joint dislocations, and easy bruising. In February 2023, he was admitted to the emergency department following a seizure, syncope, and altered sensorium, which led to an extensive evaluation of his non-specific symptoms. Ultimately, he was diagnosed with vEDS.
Keywords
Vascular Ehlers–Danlos syndrome
COL3A1 gene
Hemoptysis
Hemopneumothorax
Hypermobile joints

INTRODUCTION
Vascular Ehlers–Danlos syndrome (vEDS) is a rare and hereditary connective tissue disorder characterized by a distinct clinical presentation, including skin hyperelasticity, joint hypermobility, atrophic scarring, recurrent hemoptysis, and spontaneous pneumothorax.[1] Due to its rarity and the non-specific nature of imaging findings, early diagnosis is crucial to prevent severe consequences.[2] While clinical evaluation is essential for diagnosis, identifying the underlying genetic mutations in collagen-encoding genes or related proteins is necessary to determine the specific type or variant of Ehlers–Danlos Syndrome (EDS).[3]
CASE REPORT
A 20-year-old male presented to the emergency department in February 2023 with a seizure, followed by syncope and impaired sensorium. He had received cardiopulmonary resuscitation (CPR) from relatives before arriving at the hospital. The patient had been inconsistently taking medications for a seizure disorder over the past 2 years. Magnetic resonance imaging (MRI) and electroencephalogram results were normal. However, a chest X-ray revealed a right-sided hydropneumothorax with the right lung collapsed toward the hilum.
A contrast-enhanced computed tomography (CECT) scan of the chest conducted on February 9, 2023 [Figure 1] showed moderate to marked right-sided pneumothorax, compression of the underlying lung parenchyma, and mild mediastinal shift to the left. Additional findings included mild-to-moderate hyperdense pleural fluid and subsegmental collapse consolidation. A well-defined, thick-walled cavity with high-density fluid in the right upper lobe, accompanied by mild surrounding ground-glass opacity and plate atelectasis, was observed. Given the history of chest compressions, this finding is interpreted as lung lacerations with a hemato-pneumatocele and surrounding lung contusion. In addition, a similar but smaller air-filled cavity with dependent fluid was noted in the posterior segment of the right upper lobe, also with surrounding plate atelectasis. The left lung shows no focal lesions or pneumothorax at this time.
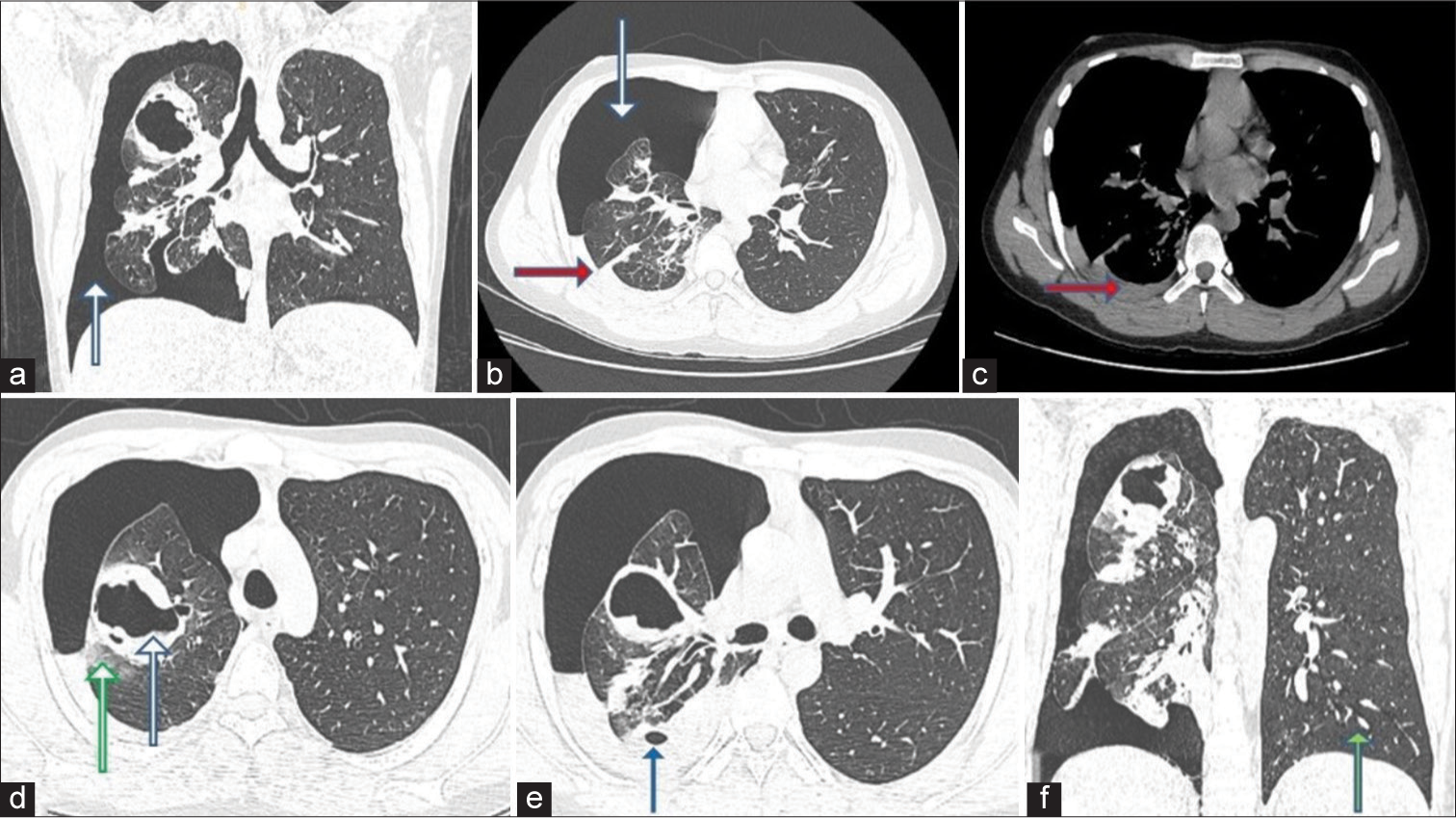
- A 20-year-old male presented with recurrent hemoptysis, recurrent knee joint dislocations, and easy bruisability of skin. Axial and coronal high-resolution computed tomography chest images taken on February 9, 2023, show (a and b) moderate-marked right-sided pneumothorax (white arrows), compression of underlying lung parenchyma (red arrow) and mild mediastinal shift to the left side. (c) Mild-moderate hyperdense pleural fluid on the right side (red arrow). There is subsegmental collapse consolidation of the adjacent right lower lobe. (d) Well, defined thick-walled cavity (blue arrow) with high-density dependent fluid in the right upper lobe with mild surrounding ground glass opacity (green arrow) and plate atelectasis. (e) Another similar but smaller air-filled cavity with dependent fluid noted in the posterior segment of the right upper lobe (blue arrow) with surrounding plate atelectasis. (f) The left lung (green arrow) shows no focal lesions/pneumothorax at this point.
The patient was treated for traumatic right hemopneumothorax with intercostal drainage, which was removed 3 days later. He was discharged after his condition improved. However, 2 months later, on April 10, 2023, he presented with acute dyspnea, cough, and several episodes of hemoptysis following a syncope and fall, for which he received CPR again.
A portable chest X-ray revealed a recurrent pneumothorax of less severity and no hydrothorax. A high-resolution computed tomography (HRCT) scan of the chest performed on April 10, 2023, [Figure 2] revealed a recurrence of hemopneumothorax. Compared to the previous scan in February, the extent of the pneumothorax has decreased. The previously identified thick-walled cavity in the apical segment of the right upper lobe has collapsed to a slit-like shape. A few of the previously observed cavities in the posterior segment of the right upper lobe are now more visible due to lung expansion and the reduction of pneumothorax. Multiple well-defined, branching, soft-tissue density nodules were noted in both lung fields. Some of these nodules are surrounded by ground-glass attenuation and were not present in the previous scan. Several of the nodules show evidence of cavitation. In addition, a few thin-walled cystic lesions were observed in the right lung, predominantly in the lower lobe.
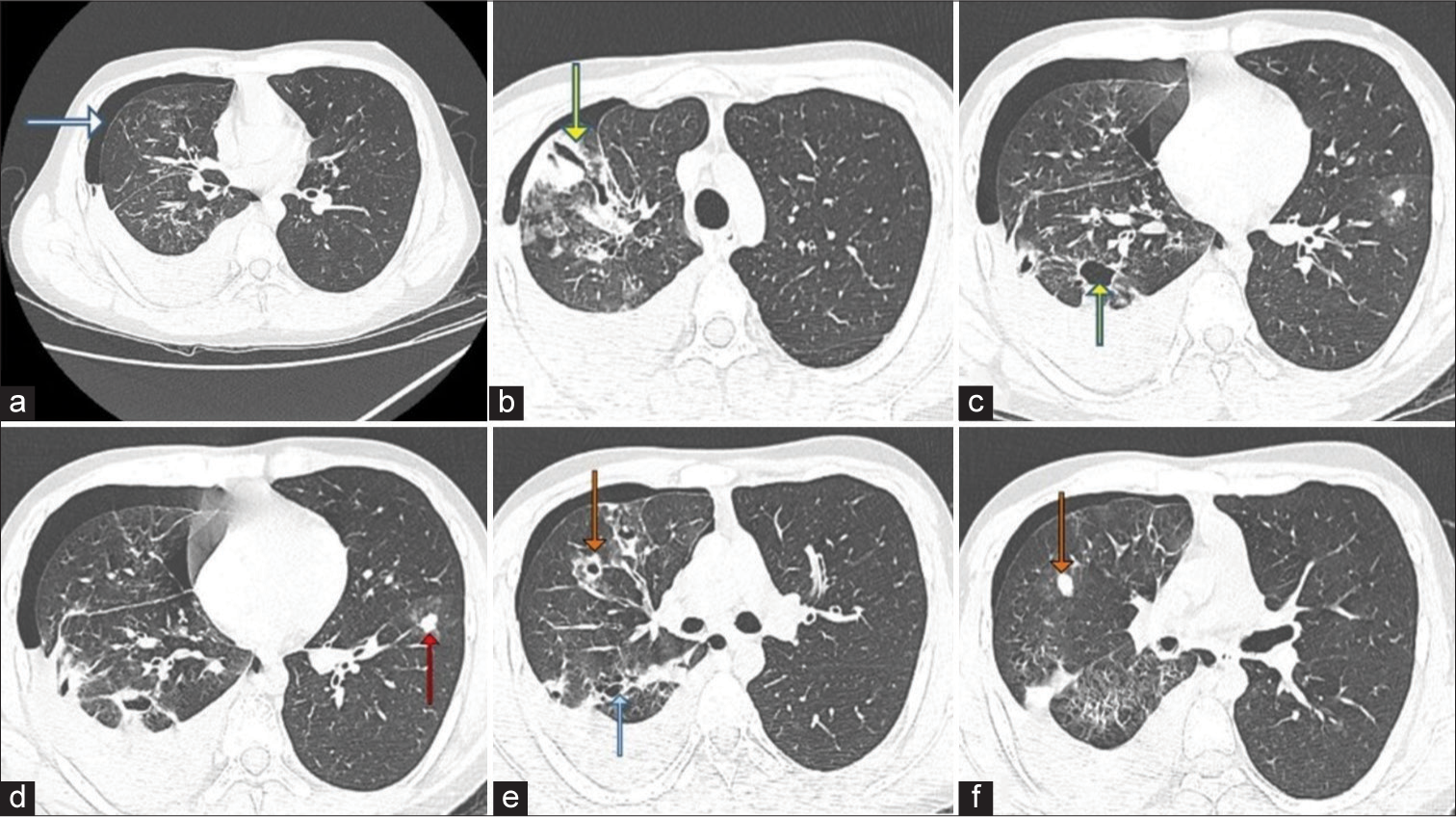
- A 20-year-old male presented with recurrent hemoptysis, recurrent knee joint dislocations and easy bruisability of skin. Axial high-resolution computed tomography chest images taken on April 10, 2023, show (a) recurrence of hemopneumothorax (white arrow). When compared to the scan of February, the extent of pneumothorax has decreased, (b) the previously mentioned thick-walled cavity (yellow arrow) in the apical segment of the right upper lobe had collapsed to a slit-like shape. (c) A couple of the previously seen cavities (yellow arrow) in the posterior segment of the right upper lobe are better seen due to the expansion of the lung and reduction of pneumothorax. (d) Multiple well-defined, branching soft-issue density nodules (red arrow) like lesions were noted involving bilateral lung fields. Some of these nodules are surrounded by ground-glass attenuation. These lesions were not seen in the previous scan. (e) Some of the nodules show evidence of cavitation (orange arrow). Few thin-walled, cystic lesions (blue arrow) in the right lung, predominantly in the lower lobe, (f) soft tissue density nodule (orange arrow).
The patient was admitted to the pulmonology department for further evaluation. Diagnostic thoracocentesis revealed blood-stained fluid, and an intercostal drainage tube was inserted. Further investigation revealed a history of mitral valve prolapse, right lung cyst, recurrent knee joint dislocations, easy bruisability, and atopy. Physical examination suggested marfanoid habitus, joint hypermobility, and positive wrist and thumb signs.
Blood routine shows a mild decrease in hemoglobin (12.3 g/dL). Bleeding time and clotting time were within normal limits. Sputum for fungal stain was negative.
Workups for tuberculosis, vasculitis, and collagen vascular diseases were negative. A third HRCT scan performed on April 18, 2023, [Figure 3] assessed the patient’s progress before discharge, revealing surgical emphysema, re-expansion of a borderline thick-walled cystic lesion, and significant resolution of ground-glass opacities bilaterally.
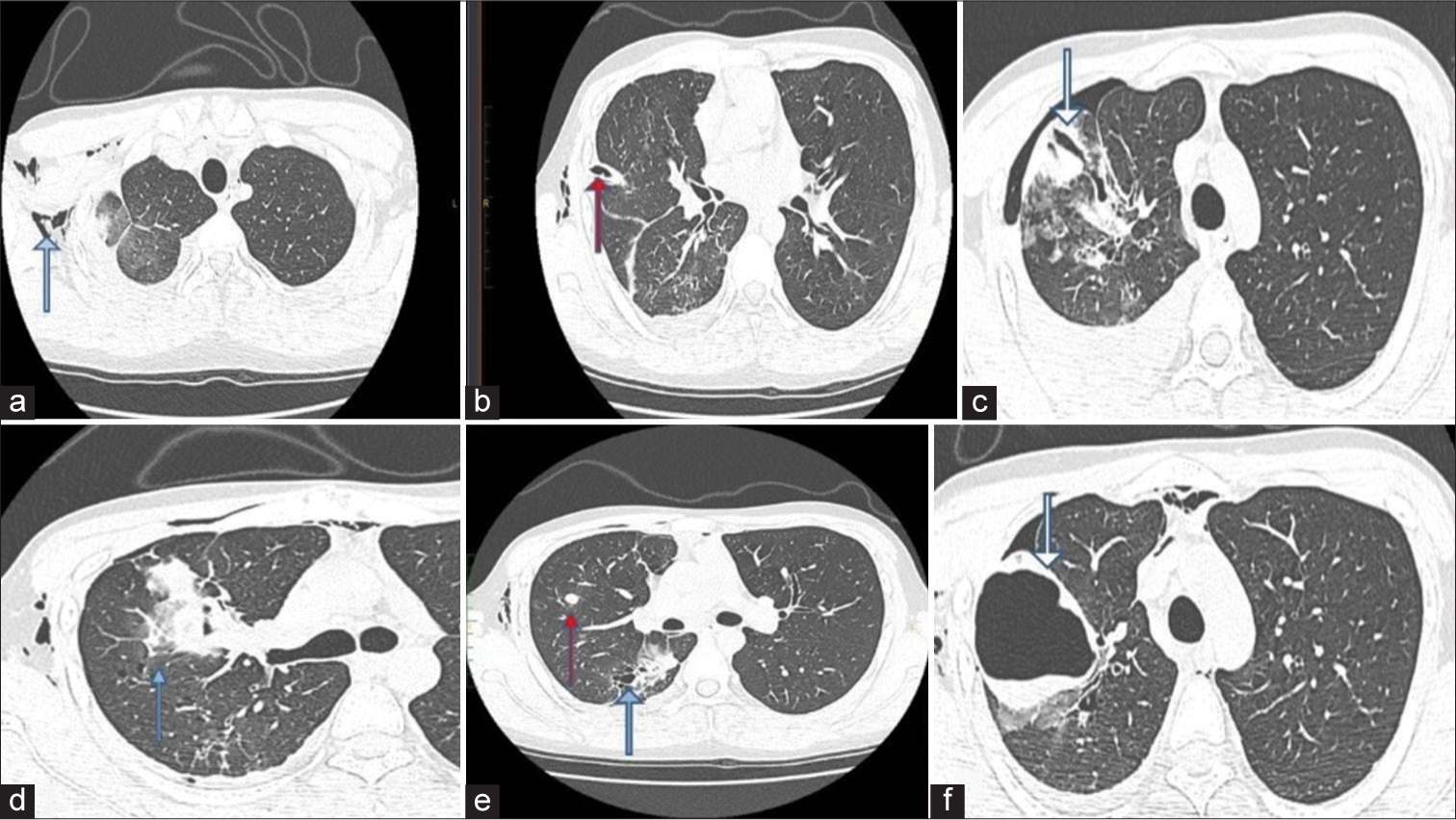
- A 20-year-old male presented with recurrent hemoptysis, recurrent knee joint dislocations and easy bruisability of skin. Axial high-resolution computed tomography chest images taken on 18/04/2023 show (a) Surgical emphysema (blue arrow) noted involving the anterior and lateral walls of the right side of the chest as well as soft tissues of the neck, (b) Interval insertion of right Intercostal drainage tube (red arrow) noted with the tip in the upper aspect of oblique fissure. (c) Showing slit-like cavity (white arrow) in the study done on 10/04/2023, however the images taken on 18/04/2023. (d) There is significant resolution in the ground glass opacities (blue arrow) bilaterally (e) Previously noted nodules (red arrow) in bilateral lung fields appear stable in size and number. Thin-walled cystic lesion (blue arrow) in the right lung persists, (f) show a re-expansion of borderline thick-walled cystic lesion (white arrow) in the apical segment of the right upper lobe, with dependent hyperdense fluid within. Pneumohemothorax has comparatively reduced.
Bronchoalveolar lavage performed the next day showed strong positivity for galactomannan. The Aspergillus skin test was 3+ for Aspergillus fumigatus. However, this did not fully explain the clinical features, particularly the recurrent hemopneumothorax and hemoptysis.
The patient was started on voriconazole on May 1, 2023, considering the possibility of a superadded fungal infection. Four months later, in August 2023, the patient was readmitted with a history of recurrent hemoptysis. A repeat HRCT chest scan performed on August 22, 2023, shows that the previously noted large cavity has been replaced by fibrotic band-like opacities. The nodules previously observed in the left lower lobe now appear to be cavitating, with surrounding ground-glass opacities. There is no evidence of hemothorax, pneumothorax, or pleural effusion. Numerous new cavitating nodules and cysts with extensive ground-glass opacities were noted in the left lower lobe, particularly in the basal regions. Whole exome sequencing revealed a likely pathogenic variant in the COL3A1 gene, confirming the diagnosis of vEDS with a superadded fungal infection. The patient underwent MRI 3D Time of flight (TOF) of the neck and circle of Willis vessels and CECT abdomen-pelvis to screen arterial territories, and no significant abnormalities detected.
The patient showed significant clinical and radiological improvement with 6 months of antifungal treatment and was advised to avoid contact sports, trauma, and physical exertion.
DISCUSSION
EDS is a collective term for a group of inherited connective tissue disorders characterized by blood vessel fragility, atrophic scarring, joint hypermobility, and skin hyperelasticity.[4] While most diagnoses are made clinically, identifying the specific type of EDS requires genetic analysis to determine the collagen or protein mutation involved.[5]
In 2017, EDS was classified into 13 variants, each associated with distinct collagen pathology.[4] The main variants include classical, vascular vEDS, hypermobile, arthrochalasis, kyphoscoliotic, and dermatosparaxis. Accurate clinical identification of EDS variants is crucial, particularly in recognizing the vascular type, which is associated with life-threatening complications such as organ perforation and arterial rupture.[6]
Vascular manifestations of vEDS include fragile blood vessels, arterial aneurysms, dissections, and dolichoectasia.[7]
Major diagnostic criteria for vEDS include:
Family history with documented COL3A1 variant
Arterial rupture at a young age
Spontaneous sigmoid colon perforation
Uterine rupture during the third trimester
Carotid-cavernous sinus fistula formation.
In adition there are 13 minor criteria, three of which were present in our patient:
Unexplained bruising in unusual sites, spontaneous pneumothorax, and hypermobility of small joints.
Our patient met one major criterion (arterial rupture at a young age, which caused hemoptysis) and three minor criteria according to the 2017 diagnostic criteria for vEDS. However, even experienced clinicians may find clinical diagnosis challenging. A formal diagnosis relies on molecular confirmation through genetic variant identification.[5] Systemic involvement and radiological features of vEDS, aside from thoracic manifestations, include: Subcutaneous calcifications,[7] ectopic ossifications,[4] skeletal manifestations such as hemarthrosis, recurrent dislocations, precocious osteoarthritis, and kyphoscoliosis.[5]
Other rare manifestations include diaphragmatic hernia, emphysema, and gastrointestinal ectasias.[6]
Effective management involves monitoring and addressing vascular and organ fragility. Patients with vEDS should modify their lifestyle, create an emergency plan, and collaborate with their care team to develop a personalized care plan.[4]
CONCLUSION
Patients presenting with a combination of recurrent pneumothorax, intrapulmonary cavities, nodular lesions, as well as skin that appears thin and transparent and joints that exhibit hypermobility, should be evaluated for vEDS. The presence of these clinical features, particularly when observed together, strongly suggests the need for further diagnostic investigation into vEDS. Early recognition and diagnosis are essential for managing this condition effectively and preventing potential complications.
TEACHING POINTS
Vascular Ehlers–Danlos syndrome (vEDS), also known as type IV Ehlers–Danlos syndrome, is the most severe form of the condition, often associated with neurovascular complications resulting from vessel dissections and/or aneurysms
This syndrome is caused by a mutation in the COL3A1 gene, which codes for type III procollagen, leading to fragile connective tissues and an increased risk of arterial, gastrointestinal, and uterine ruptures.
MCQs
-
Which of the following is NOT a major criterion for the diagnosis of vascular Ehlers–Danlos syndrome (vEDS)?
Family history of vEDS with documented causative variant in COL3A1
Arterial rupture at a young age
Spontaneous sigmoid colon perforation in the absence of known diverticular disease or other bowel pathology
Spontaneous pneumothorax
Answer Key: d (This is a minor criterion, not a major one)
-
Heterozygous mutations in which gene result in vascular Ehlers–Danlos syndrome (vEDS)?
COL3A1 gene
COL2A1 gene
COL3A2 gene
COL2A2 gene
Answer Key: a
-
Systemic involvement in vascular Ehlers–Danlos syndrome (vEDS) includes all of the following EXCEPT:
Spontaneous dislocation of the temporomandibular joint
Subcutaneous calcifications
Ectasias of the gastrointestinal tract
Pathological fractures
Answer Key: d (This is not a typical feature of vEDS)
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Vascular Ehlers-Danlos syndrome: Clinical presentation and diagnosis. Am J Med Genet. 2020;182:10-20.
- [Google Scholar]
- Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017;175:40-7.
- [CrossRef] [PubMed] [Google Scholar]
- Ehlers-Danlos syndrome type IV In: GeneReviews. 2015. Available from: https://www.ncbi.nlm.nih.gov/books/NBK47426 [Last accessed on 2024 Jul 03]
- [Google Scholar]
- The spectrum, management, and clinical outcome of Ehlers-Danlos syndrome type IV: A 30-year experience. J Vasc Surg. 2005;42:98-106.
- [CrossRef] [PubMed] [Google Scholar]
- Vascular Ehlers-Danlos syndrome In: GeneReviews. 2014. Available from: https://www.ncbi.nlm.nih.gov/books/NBK47427 [Last accessed on 2024 Jul 03]
- [Google Scholar]




